Benchmark Dataset for Catalysis on 2D MXenes
May 30, 2026·
 ,
,
,
,
,
,
·
2 min read
,
,
,
,
,
,
·
2 min read
Pavlo Melnyk*
Anmar Karmush*
Mårten Wadenbäck
Ania Beatriz Rodríguez-Barrera
Johanna Rosen
Michael Felsberg
Jonas Björk*
Abstract
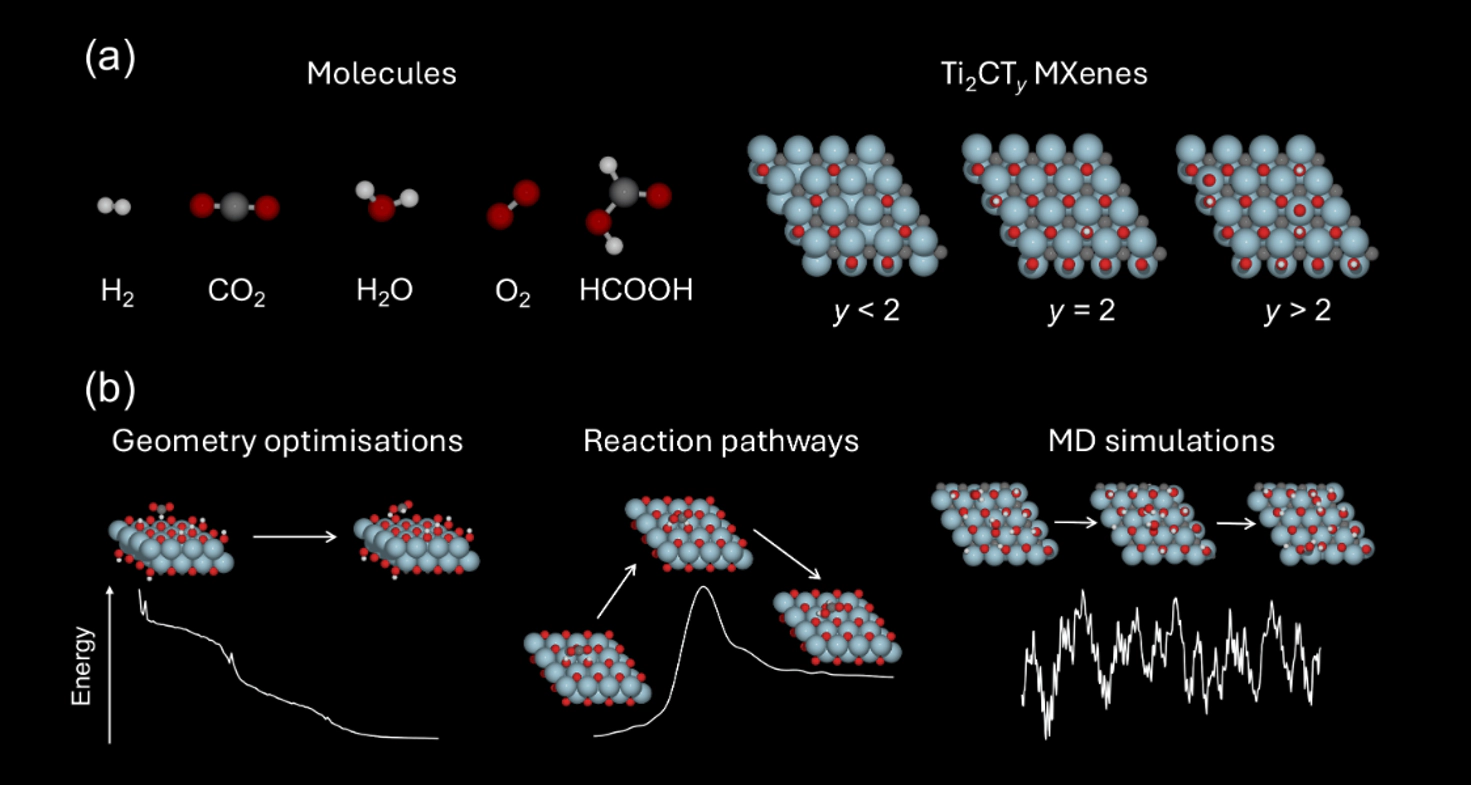
Merging first-principles calculations with machine learning (ML), we aim to accelerate the exploration of catalytic behaviour in novel materials. We focus on two-dimensional (2D) Ti2CTy MXenes, whose versatile surface chemistry makes them particularly compelling candidates for catalysis. Resolving their composition and structure under realistic conditions exceeds the reach of standard density functional theory (DFT) due to computational cost.
To address this challenge, we generate a comprehensive dataset of 50,000 DFT calculations for training and 10,000 for testing, encompassing both Ti2CTy MXene configurations and molecular systems, along with an additional test dataset with 1000 genuinely new, larger systems to investigate how well models generalise. We train and validate widely used and competitive machine learning interatomic potentials (MLIP) models, EquiformerV2, MACE, MatRIS, UPET, and MatRIS that accurately predict atomic forces and formation energies — quantities that DFT must repeatedly compute for structural and catalytic investigations — for these 2D materials.
This combined DFT–ML framework achieves computational acceleration of the order ∼1 − 4 · 10$^3$ (on a CPU) while maintaining desired-level accuracy (∼±10 meV/A for forces and ∼±1 meV for per-atom energies), paving the way for more efficient investigations of MXene catalytic behaviour. Moreover, we perform an extensive qualitative evaluation of the trained models, showcasing the importance of the comprehensive simulation-based comparison beyond the benchmark metrics. The dataset and the trained models with the code are available at https://huggingface.co/datasets/CatalystAnonymous/catalyst_mxenes.
To address this challenge, we generate a comprehensive dataset of 50,000 DFT calculations for training and 10,000 for testing, encompassing both Ti2CTy MXene configurations and molecular systems, along with an additional test dataset with 1000 genuinely new, larger systems to investigate how well models generalise. We train and validate widely used and competitive machine learning interatomic potentials (MLIP) models, EquiformerV2, MACE, MatRIS, UPET, and MatRIS that accurately predict atomic forces and formation energies — quantities that DFT must repeatedly compute for structural and catalytic investigations — for these 2D materials.
This combined DFT–ML framework achieves computational acceleration of the order ∼1 − 4 · 10$^3$ (on a CPU) while maintaining desired-level accuracy (∼±10 meV/A for forces and ∼±1 meV for per-atom energies), paving the way for more efficient investigations of MXene catalytic behaviour. Moreover, we perform an extensive qualitative evaluation of the trained models, showcasing the importance of the comprehensive simulation-based comparison beyond the benchmark metrics. The dataset and the trained models with the code are available at https://huggingface.co/datasets/CatalystAnonymous/catalyst_mxenes.
Type
Publication
In arXiv