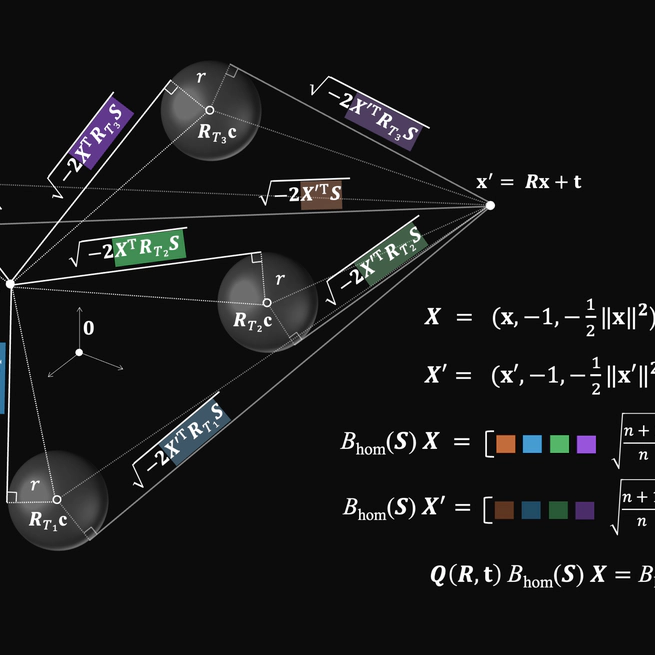
E$(n)$-Equivariant Spherical Decision Surfaces
We present a constructive derivation of exactly E(n)-equivariant spherical decision surfaces by extending prior O(n)-equivariant hypersphere neurons to include translations. To achieve this, we present a decomposition of the features of the O(n)-equivariant neurons and provide explicit representations for translation and E(n)-transformations to fulfil the respective equivariance constraints. The resulting decision surfaces are exactly E(n)-equivariant without input centring or explicit pairwise differences, and admit explicit closed-form matrix representations. In addition, we numerically verify the correctness of the derivations and perform a downstream check of the resulting geometric primitives.
Mar 2, 2026
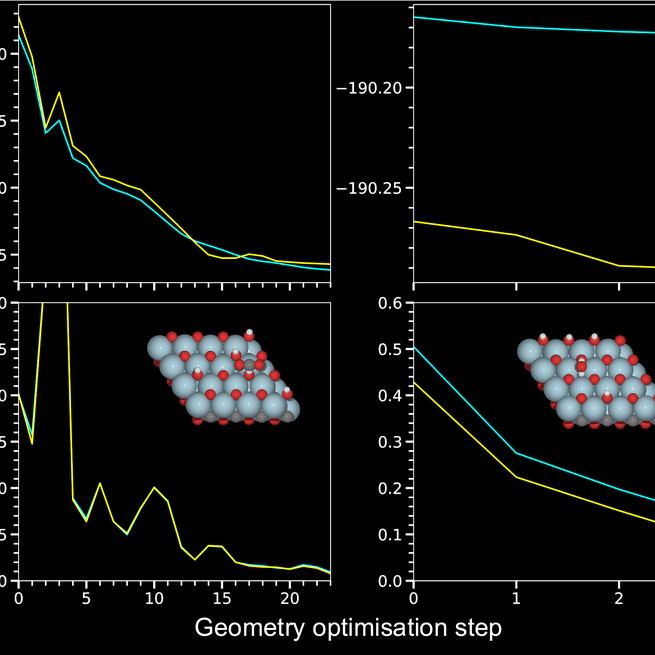
Equivariant Modelling for Catalysis on 2D MXenes
Merging advanced computations with machine learning, we aim to accelerate the exploration of catalytic behaviour in novel materials. We focus on two-dimensional (2D) Ti$_2$CT$_y$ MXenes, whose versatile surface chemistry makes them particularly compelling candidates for catalysis. However, resolving their composition and structure under realistic conditions requires going beyond the systems typically studied with density functional theory (DFT), as the computational cost of such calculations limits accessible system sizes and timescales, calling instead for more efficient approaches. To address this challenge, we generate a comprehensive dataset of 50,000 DFT calculations for training and 10,000 for testing, encompassing both Ti$_2$CT$_y$ MXene configurations and molecular systems, along with an augmented dataset where systems are artificially repeated to investigate how well models generalise to larger systems.Employing advances in geometric deep learning, we train and validate an equivariant (\ie symmetry-aware) model (EquiformerV2) that accurately predicts atomic forces and formation energies — quantities that DFT must repeatedly compute for structural and catalytic investigations — for these 2D materials. This combined DFT–ML framework achieves computational acceleration of the order ${\sim}10^3$–$10^4$ (on a CPU) while maintaining DFT-level accuracy (${\sim} {\pm} 45$ meV/Å for forces and ${\sim} {\pm} 6$ meV for per-atom energies), paving the way for more efficient investigations of MXene catalytic behaviour. Moreover, we confirm that the total energy prediction error of the model grows linearly with the number of atoms in an input system, while the force error remains the same, which, along with the equivariant model design, is a necessity for a robust model. The dataset and the trained models with the code are available at \url{https://github.com/CataLiUst}.
Nov 24, 2025